Class 17: Genome Assembly
Bioinformatics
Andrés Aravena
December 09, 2021
Base calling
From chromatograms to sequences
Sequencers produce signals
- Sanger produces chromatograms
- Pyrosequencing make videos
- others
each method has their own tool to call the bases
For Sanger, that tool is called Phred
Phred

Phred quality
Signal–Ratio is different for each base
As usual we use logarithms, multiply by a constant, and take the integer part \[\lfloor10\cdot\log_{10}(\text{Signal}/\text{Noise})\rfloor\]
Phred quality
| Q | Error.probability | Probability.of.correct |
|---|---|---|
| 10 | 0.1000000 | 0.9000000 |
| 15 | 0.0316228 | 0.9683772 |
| 20 | 0.0100000 | 0.9900000 |
| 30 | 0.0010000 | 0.9990000 |
| 40 | 0.0001000 | 0.9999000 |
| 50 | 0.0000100 | 0.9999900 |
| 60 | 0.0000010 | 0.9999990 |
| 70 | 0.0000001 | 0.9999999 |
| 80 | 0.0000000 | 1.0000000 |
Phred quality of each base

Phred file format (.phd)
BEGIN_SEQUENCE GTFAG75TF.scf
BEGIN_COMMENT
CHROMAT_FILE: GTFAG75TF.scf
ABI_THUMBPRINT: 0
TRACE_PROCESSOR_VERSION:
BASECALLER_VERSION: phred 0.071220.c
PHRED_VERSION: 0.071220.c
CALL_METHOD: phred
QUALITY_LEVELS: 99
TIME: Fri Nov 22 15:00:37 2019
TRACE_ARRAY_MIN_INDEX: 0
TRACE_ARRAY_MAX_INDEX: 9116
TRIM: 34 555 0.0500
TRACE_PEAK_AREA_RATIO: 0.0255
CHEM: prim
DYE: rhod
END_COMMENT
BEGIN_DNA
g 6 8
a 6 24
g 6 28
a 8 45
g 8 50
a 8 59
t 8 77
c 8 87
t 9 100
a 9 115
g 11 124
t 11 140
g 15 147
t 8 162
g 9 170
c 12 182
t 9 197
g 17 207
g 20 221
a 21 234Files of type .qual
This is FASTA format, with numbers instead of letters
Each number is the quality of the corresponding letter
>GTFAG75TF.scf 702 0 702 SCF
6 6 6 8 8 8 8 8 9 9 11 11 15 8 9 12 9 17 20 21 20 18 9 10 6 6 6 6 8 16 11 6 6 8
20 17 30 30 44 44 29 16 9 9 9 22 23 44 44 32 32 32 34 34 34 38 38 38 38 34 38 38
38 38 44 34 34 34 39 44 47 47 53 53 53 53 53 47 47 47 47 47 47 53 53 47 53 53 53
47 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59
59 59 59 59 59 24 24 59 59 59 59 62 62 62 62 62 62 62 59 59 59 59 59 59 51 59 59
62 59 59 59 59 59 59 59 59 56 59 59 59 59 59 56 56 56 39 38 38 38 56 56 56 56 59
62 62 62 62 59 59 59 59 59 59 59 62 62 62 62 62 62 44 40 40 28 28 28 59 59 56 59
59 56 56 56 56 59 56 56 59 59 59 59 59 59 59 59 56 56 56 56 56 56 59 59 59 59 62
59 59 56 56 56 59 56 59 59 59 59 59 59 47 59 59 59 59 59 59 59 59 59 59 59 59 59
56 56 56 56 56 59 62 59 59 59 59 59 59 59 59 59 59 59 59 62 62 62 62 59 59 59 59
59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 59 25 25 59 59 59 59 59 59
59 59 59 59 59 59 59 59 59 59 53 53 43 43 43 53 53 53 53 53 53 53 53 53 53 47 47
47 47 47 44 39 53 39 39 39 39 39 53 53 53 53 53 47 47 43 44 38 38 32 38 27 27 32
31 29 29 35 35 36 35 35 38 38 43 43 43 38 53 39 34 34 53 53 47 43 47 53 53 53 53
53 53 47 47 43 39 32 32 29 30 30 38 34 35 26 44 44 44 38 33 26 24 27 29 33 33 35
38 38 38 34 34 34 38 32 38 34 34 34 39 32 39 34 30 32 32 34 34 38 38 38 38 38 38
38 38 29 29 27 18 17 27 27 27 20 20 33 29 33 32 32 32 35 38 38 34 34 34 35 47 34
38 32 32 25 27 20 20 15 24 30 32 32 32 31 29 29 27 29 32 31 24 29 33 32 33 31 28
29 29 24 24 29 29 29 32 32 31 29 24 29 24 24 23 19 20 23 21 21 17 18 10 10 10 9
7 9 9 9 9 11 19 25 21 21 20 23 24 24 24 18 18 10 10 10 18 20 20 14 24 15 20 20
29 24 17 20 15 14 14 12 11 9 8 6 6 6 8 8 8 10 10 10 18 18 24 24 16 18 13 11 11
14 10 9 16 11 10 10 11 10 13 10 19 21 17 8 6 6 6 11 10 11 11 10 10 12 10 19 18 9
7 10 6 6 6 9 11 9 10 10 12 8 6 6 6 6 8 8 9 9 9 9 8 11 11 11 7 7 11 6 6 6 6 6 6 9
7 6 6 8 8 11 6 6 6 6 6 6 6 9 6 6 6 8 6 8 9 8 6 6 6 6 6 9 6 6 8 6 6 6 6 6 8 11 12
11 9 9 9 7 12 8 9 7 9 9 9 6 6 9 9 7 7 7 7 FASTQ
Inspired by FASTA, combining sequences and qualities
- Each entry starts with
@instead of>, and the read ID - The second line is the sequence, base by base
- Line 3 starts with
+, can be followed by the ID again - Line 4 has the qualities encoded, one symbol for each base
@001043_1783_0863
CTGATCATTGGGCGTAAAGAGTGCGCAGGCGGTTTGTTAAGCGAGATGTGAAAGCCCCGGGCTCAACCTGGGAATTGCATTTCGAACTGGCGAACTAGAGTCTTGTAGAGGGGGTAGAATTCCAGGTGTAGCGGTGAAATGCGTAGAGATCTGGAGGAATACCGGTGGCGAAGGCGGCCCCCTGGACAAAGACTGACGCTCAGGCACGAAAGCGTGGGGAGCAAACAGGATTAAATACCCTCGTA
+001043_1783_0863
<<<<<<<>;>=0<<<=<-<<<<<<<<<>:<>:>=/<?;?;<<<<<<<<<<>=0<<<5(=<-<<<>;?;<>=0>;?;<<<>=/<<>:;<<8<<?;<<<:<<<<?;<<<<<;:71(;;;?;>9>:<>:;<<<;<>9<<>=1<<<<<<<<<<<<<>;;?;>:<:?;?;<>:<<?;?;<?;;;70';>;9<>=1;:<;;;:<<<<<?;;<<<>=2<<;<<<5(<;9>=0;<?;<>;=<2:8>=1:9<<9Encoding quality in a single letter
Each quality value between 0 and 90 is represented by the corresponding symbol in the following text
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~Initially FASTQ were supposed to have sequences and qualities in several lines, but the symbols + and @ can be quality values
To avoid ambiguity, each entry has exactly 4 lines.
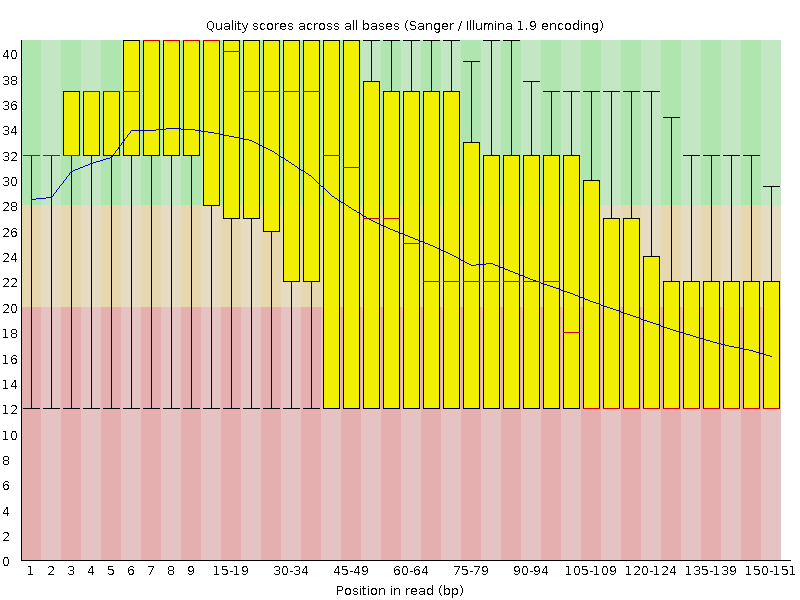
Quality control: FastQC
Assembly
Assembling genomes
Once we have all the reads, how do we get the genome?
Imagine a book of 1000 pages:
- several copies of the book are cut randomly in one million pieces
- different pieces may overlap
- half of the pieces are lost
- the remaining half is splashed with ink in the middle
The problem is to reconstruct the original book
When not to assemble
If we already know the genome, we do not need to assemble it
This is obvious. Still, it is an important case
- When we sequence expressed genes, i.e. RNAseq
- When we look for polymorphisms
De novo Strategies
Also called ab initio
These are Latin words meaning “from new” or “from the start”
There are basically three strategies used to assemble genomes
- Greedy algorithms
- Overlap – Layout – Consensus
- De Bruijn graphs
Today we will speak about the first two strategies
Greedy algorithm
Given a set of sequence fragments, the object is to find a longer sequence that contains all the fragments.
- Evaluate the pairwise alignments of all fragments
- Choose two fragments with the largest overlap
- Merge the chosen fragments
- Repeat step 2 and 3 until only one fragment is left.
Overlap
- All reads are compared to each other
- and to their reverse-complement
- The alignment considers each base’s quality
- When the end of one read matches the start of another read, we say that they overlap
Sometimes reads do overlap

Sometimes they do not overlap

Problems with the greedy approach
The final sequence may be wrong
Two different reads A, B may match the same read X
To decide which one is correct, a Layout stage is used
Layout
In this approach, the overlap between reads is used to build a graph of reads relationships
This graph determines the layout of all reads,
that is, their relative positions
Contigs
Usually there are several independent groups of reads that are not connected in the layout graph
(we say that the graph has several connected components)
All the reads that are connected together form a contig
Consensus
Once the layout is clear, the last step is to retrieve the consensus sequence of the contig
In fact, it is not a consensus. It is a vote. The majority wins
High quality votes are more important than low quality ones
Example: Phrap assembler
The human genome project used Phred for base-calling and Phrap for assembly
Phrap produces several files. The most important has extension .ace
These programs are free for academic usage (non-commercial)
Example ACE file
AS 36 39
CO Contig1| 131 1 1 U
gctagaaaaaaaaggactcccagtagaaatacgtacaataaagtaggttc
ctctagttaactgttacaaaataagtttcccattggtaatataatagatt
tataactgttatatccagagcaacctagggg
BQ
15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15
15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15
15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15
15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15
15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15 15
15
AF 5765593 U 1
BS 1 131 5765593
RD 5765593 131 0 6
gctagaaaaaaaaggactcccagtagaaatacgtacaataaagtaggttc
ctctagttaactgttacaaaataagtttcccattggtaatataatagatt
tataactgttatatccagagcaacctaggggExample ACE file (last part)
CO Contig36 111 2 63 U
taTAAAGTCGATGGGGAGGAAGATAGGGGAGCTAAAGCCATAGGGAAACC
ACGTAGTTCTGCGTCAAGCGTTgccttcCGAGGTGCTCTCCGCTTTTCCA
TGCtccaatcg
BQ
15 15 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25
25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25
25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 15 15 15 15 15 15
25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 25 15
15 15 15 15 15 15 15
AF 5648323 U 1
AF 5703145 U 1Depth

Online Material
Indiana University Bloomington, “Introduction to Bioinformatics”, Lecture by Yuzhen Ye
Université Lyon I, Network Algorithms for Molecular Biology lesson on “Introduction to (de novo) assembly”, by Blerina Sinaimeri
MIT, “Foundations of Computational Systems Biology”, Lecture by David K. Gifford