Class 17: Understanding SAM files
Bioinformatics
Andrés Aravena
December 9, 2022
We can align reads to a genome
There are several tools used to map reads to a genome
bwabowtiebowtie2hisathisat2
Each one has several options, that you can read in their manual
Output format
All these tools produce the same kind of files
Sequence Alignment/Mapping (SAM) files are text files
They are big. Sometimes they are encoded in binary
BAM files are Binary SAM files
Humans and all programs can read text files, only some programs can read binary files
What is inside the .sam file?
@SQ SN:XI LN:666816
@SQ SN:IX LN:439888
@SQ SN:IV LN:1531933
@SQ SN:III LN:316620
@SQ SN:X LN:745751
@SQ SN:XII LN:1078177
@SQ SN:VIII LN:562643
@SQ SN:VI LN:270161
@SQ SN:V LN:576874
@SQ SN:II LN:813184
@SQ SN:XIV LN:784333
@SQ SN:XIII LN:924431
@SQ SN:XVI LN:948066
@SQ SN:XV LN:1091291
@SQ SN:I LN:230218
@SQ SN:Mito LN:85779
@SQ SN:VII LN:1090940
@PG ID:bwa PN:bwa VN:0.7.17-r1188 CL:bwa mem yeast.fasta y1.fastq -o out1.sam
SRR507778.1 0 Mito 20158 60 36M * 0 0 ANTATAATATTATCCCCACGAGGGCCACACATGTGT ?#?;<BBB?BBGGEGGGGDDE8EEDB:?<=?BB?B7 NM:i:1 MD:Z:1A34 AS:i:34 XS:i:0
SRR507778.2 16 XV 767197 60 36M * 0 0 ACCCATTTCATAATTAATAAGTGGTATATCAGGANG <?DGEDBHHHHHHHDHFGFHFGFGHG@@A=@;;7#: NM:i:1 MD:Z:34C1 AS:i:34 XS:i:0
SRR507778.3 0 II 569438 60 36M * 0 0 GNAAGAACCCTTATACTAACAGCAGCATAGGAAAGT ;#>45<4454GEDGGGGGGECEACBBBB=FGADAG8 NM:i:1 MD:Z:1C34 AS:i:34 XS:i:0What does this mean?
Lines starting with @ are metadata
They describe the conditions of the alignment
Line like this
describe the sequence
What does that mean?
Line like this
describe how the program was executed. It includes
- ID: id of the command
- PN: program name
- VN: version number
- CL: command line
What is the alignment data?
Many lines like this:
SRR507778.1 0 Mito 20158 60 36M * 0 0
ANTATAATATTATCCCCACGAGGGCCACACATGTGT
?#?;<BBB?BBGGEGGGGDDE8EEDB:?<=?BB?B7
NM:i:1 MD:Z:1A34 AS:i:34 XS:i:0
SRR507778.2 16 XV 767197 60 36M * 0 0
ACCCATTTCATAATTAATAAGTGGTATATCAGGANG
<?DGEDBHHHHHHHDHFGFHFGFGHG@@A=@;;7#:
NM:i:1 MD:Z:34C1 AS:i:34 XS:i:0
SRR507778.3 0 II 569438 60 36M * 0 0
GNAAGAACCCTTATACTAACAGCAGCATAGGAAAGT
;#>45<4454GEDGGGGGGECEACBBBB=FGADAG8
NM:i:1 MD:Z:1C34 AS:i:34 XS:i:0(these are three long lines, written in several lines)
What is the alignment data?
Each column in Sequence Alignment/Map (SAM) has a different meaning
| Column | Name | Description |
|---|---|---|
| 1 | QNAME | Query template/pair NAME |
| 2 | FLAG | bitwise FLAG |
| 3 | RNAME | Reference sequence NAME |
| 4 | POS | 1-based leftmost POSition/coordinate of clipped sequence |
| 5 | MAPQ | MAPping Quality (Phred-scaled) |
| 6 | CIGAR | extended CIGAR string |
| 7 | MRNM | Mate Reference sequence NaMe (= if same as RNAME) |
| 8 | MPOS | 1-based Mate POSition |
| 9 | TLEN | inferred Template LENgth (insert size) |
| 10 | SEQ | query SEQuence on the same strand as the reference |
| 11 | QUAL | query QUALity (ASCII-33 gives the Phred base quality) |
| 12+ | OPT | variable OPTional fields in the format
TAG:VTYPE:VALUE |
What is the flag?
Each bit in the FLAG field is defined as:
| Value | Code | Meaning |
|---|---|---|
| 1 | p | the read is paired in sequencing |
| 2 | P | the read is mapped in a proper pair |
| 4 | u | the query sequence itself is unmapped |
| 8 | U | the mate is unmapped |
| 16 | r | strand of the query (1 for reverse) |
| 32 | R | strand of the mate |
| 64 | 1 | the read is the first read in a pair |
| 128 | 2 | the read is the second read in a pair |
| 256 | s | the alignment is not primary |
| 512 | f | the read fails platform/vendor quality checks |
| 1024 | d | the read is either a PCR or an optical duplicate |
| 2048 | S | the alignment is supplementary |
What is a bit flag?
The FLAG column is a number
It is the sum of numbers on the previous table
For example
The first read is not paired, and aligned in the Forward strand
The second read is not paired, and aligned in the Reverse strand
Why there are only 0 and 16 ?
Most of the flags only make sense when we combine forward and reverse read files
Now FLAGS is more interesting
99 = 64+32+2+1 :
- 1 : the read is paired in sequencing
- 2 : the read is mapped in a proper pair
- 32 : strand of the mate
- 64 : the read is the first read in a pair
147 = 128+16+2+1 :
- 1 : the read is paired in sequencing
- 2 : the read is mapped in a proper pair
- 16 : reverse strand
- 128 : the read is the second read in a pair
What do we mean by “Paired”?
Each read has its “pair”
fragment ========================================
SE read --------->
PE reads R1---------> <---------R2
unknown gap ....................- SE
- Single end read
- PE
- Paired end read
How to know which reads are paired?
The two FASTQ files need to have the same number of reads
The first read of one file matches the first read of the other file
The n-th read of one file matches the n-th read of the other file
What do we mean by “proper paired”?
Basically
- Both pairs are in the same chromosome
- both pairs are in opposite strands
- both pairs are facing each other
Now FLAGS is more interesting
83 = 64+16+2+1 :
- 1 : the read is paired in sequencing
- 2 : the read is mapped in a proper pair
- 16 : reverse strand
- 64 : the read is the first read in a pair
163 = 128+32+2+1 :
- 1 : the read is paired in sequencing
- 2 : the read is mapped in a proper pair
- 32 : strand of the mate
- 128 : the read is the second read in a pair
CIGAR
The column called CIGAR (Concise Idiosyncratic Gapped Alignment Report) describes how the read mapped to the genome
In other words, it shows which operations are necessary to map the read to the reference
For full details see
Heng Li, Bob Handsaker, Alec Wysoker, Tim Fennell, Jue Ruan, Nils Homer, Gabor Marth, Goncalo Abecasis, Richard Durbin, 1000 Genome Project Data Processing Subgroup, “The Sequence Alignment/Map format and SAMtools”, Bioinformatics, Volume 25, Issue 16, 15 August 2009, Pages 2078–2079, https://doi.org/10.1093/bioinformatics/btp352
Operations encoded in CIGAR format
| M | Alignment (can be a sequence match or mismatch!) |
| I | Insertion in the read compared to the reference |
| D | Deletion in the read compared to the reference |
| N | Skipped region from the reference. An intron (in mRNA) |
| S | Soft clipping (clipped sequences are present in read) |
| H | Hard clipping (clipped sequences are NOT present in the alignment record); |
| P | Padding (silent deletion from padded reference) = Sequence match |
| X | Sequence mismatch (not widely used) |
Example

Interpretation
r001 is the name of a read pair. It has FLAG 163 (=1 + 2 + 32 + 128)
- It mapped to position 7 is the second read in the pair (128) and regarded as properly paired (1 + 2);
- its mate is mapped to 37 on the reverse strand (32).
Read r002 has three soft-clipped (unaligned) bases
- The coordinate shown in SAM is the position of the first aligned base.
- The CIGAR string for this alignment contains a P (padding) operation which correctly aligns the inserted sequences.
Interpretation (cont)
The last six bases of read r003 map to position 9, and the first five to position 29 on the reverse strand.
The hard clipping operation H indicates that the clipped sequence is not present in the sequence field. The NM tag gives the number of mismatches.
Read r004 is aligned across an intron, indicated by the N operation.
Another example
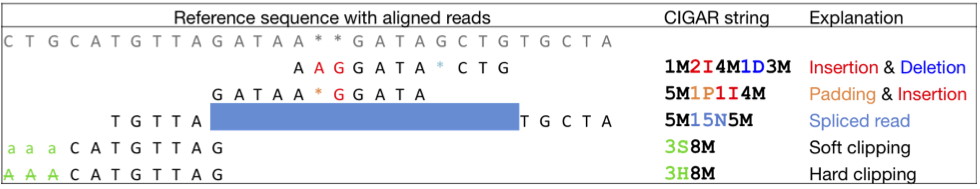
What do you see here?
How to do your own alignments?
Use Galaxy
If you have a good super-computer, and you know how to use it, then you can do it easily
For small projects it is better to use galaxy